Implementation description¶
Note
This implementation for the moment only deals with encoding and formatting the payload for the image data. It does not support the formatting of the variables necessary for decoding (frequency values of the DCT and size of the image). What can be recommended for the moment is to use a common general frequency table for the DCT values.
The jpgedna package has been implemented in the most modular way possible. We aim at making a code that people will be able to experiment with.
Coders¶
Principle¶
The main purpose of the jpegdna algorithm is to encode the DCT coefficients into quaternary code instead of binary. To achieve this, we use a series of coders that are the core of our implementation. Since these coders play such an important role, we have decided to orient the development around them. We defined a standard that all of our coders follow:
Inherit from the abstract class
coders.AbstractCoderImplement the abstract methods
mycoder.encode(inp)andmycoder.decode(code)Both those methods only communicate with the user, the data to be encoded or decoded and the data result
Additionnal arguments can be set or accessed by implementing the
mycoder.set_state(*args)andmycoder.get_state()metodsThose arguments will be defined as attributes,
mycoder.set_state(*args)andmycoder.get_state()are a setter and a getterThe
mycoder.full_encode()ormycoder.full_decode()methods can be overloaded to describe the full encoding or decoding process (initialisation, encoding, return of arguments)
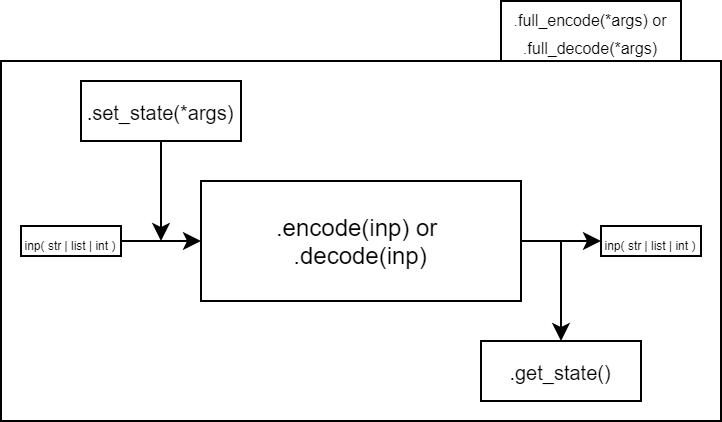
To implement a new coder, following these requirements will suffice and render the use of the coder easy. The encode and decode methods quickly help to define the payload of the data that you encode, and the getters and setters help with identifying what might be needed in some headers for the well funcitonning of the whole coder.
The coders that have already been implemented are the following:
Goldman Coder¶
The Goldman Coder is instantiated with the alphabet that will be in use for the coder: coder = jpegdna.coders.GoldmanCoder(alphabet).
The coder uses strings as input and output and does not need any initialization.
A wrapper coder = jpegdna.coders.GoldmanCoder(alphabet) has been made for the specific case where we use DNA.
- class jpegdna.coders.goldmancoder.GoldmanCoder(alphabet, verbose=False)[source]
Goldman coder
- Parameters
alphabet (list) – alphabet for encoding
- Return type
None
- decode(code)[source]
Function that decodes DNA data using Goldman coder
- Parameters
code (str) – Signal to be decoded
- Returns
Decoded signal
- Return type
str
- encode(inp)[source]
Function that encodes data into DNA using Goldman coder
- Parameters
inp (str) – Signal to be encoded
- Returns
Encoded signal
- Return type
str
Huffman Coder¶
The Huffman n-ary coder is instanciated with 3 parameters: the alphabet on which it is defined, the frequencies of appearance for the alphabet members,
and the integer describing the arity of the tree.
coder = jpegdna.coders.HuffmanCoder(alphabet, freqs, arity)
The encoder takes a list of alphabet members as input, and outputs a string of integers between 0 and n-1, n being the arity of the tree. The decoder inverses the types for input and output.
- class jpegdna.coders.huffmancoder.HuffmanCoder(*args, verbose=False)[source]
Huffman n-ary tree coder
case 1:
- Parameters
alphabet (list) – List of elements that can be encoded
feqs – List of appearence frequencies for each element in the alphabet
n (int) – base of the n-ary tree
verbose – bool
debug – bool
- Variables
verbose – Verbosity enabler
debug – Verbosity for debug enabler
case 2:
- Parameters
dic (dict) – Huffman dictionnary
- decode(code)[source]
Decode a signal using the Huffman n-ary dictionnary
- Parameters
code (str) – Signal to be decoded
- Returns
Decoded signal
- Return type
list
- encode(inp)[source]
Encode a signal using a Huffman n-ary dictionnary
- Parameters
inp (list) – Signal to be encoded
- Returns
Encoded signal
- Return type
str
- find_codeword_key(word)[source]
Find the symbol associated with a codeword in the huffman dictionnary
- Parameters
word (str) – codeword for which we want the symbol
- Returns
Symbol in the alphabet corresonding to the codeword
- Return type
str
Coefficient coder¶
The coefficient coder is the part that takes care of encoding the coefficient sequence obtained after quantizing the DCT coefficients. There are two different ones, one that is used for DC coefficients and one for AC coefficients only.
The DC coefficient is first diferrentially coded, meaning instead of encoding the coefficient, we encode the difference between itself and the DC coefficient of the previous block. Then the difference result is encoded using a combination of the category coder and the value coder. The category of a coefficient is an integer that is related to the absolute value of the coefficient. In the case where we use the paircode algorithm developed by Melpomeni Dimopoulou, in function of the relation table between the values and their category is:
Category |
Negative coefficients |
Positive coefficients |
1 |
[-5, -1] |
[1, 5] |
2 |
[-25, -6] |
[6, 25] |
3 |
[-75, -26] |
[26, 75] |
4 |
[-275, -76] |
[76, 275] |
5 |
[-775, -276] |
[276, 775] |
6 |
[-2775, -776] |
[776, 2775] |
7 |
[-7775, -2776] |
[2776, 7775] |
8 |
[-27775, -7776] |
[7776, 27775] |
The category coder will basically encode this category into a DNA word using a dictionnary of codewords for each possible category. Introducing a concept of category for the coefficients is very helpful beacause it can help with allocating an appropriate number of nucleotides necessary to encode the real value of the coefficient. This is the role of the value coder.
An important part of the coefficient decoder is the get_state() method because it gives back a value called num_bits that essentially describes the number of bits used to encode the current coefficient. That way, when you decode the coefficient, you can resynchronize your position in the quaternary stream once it is done.
Category Coder¶
The category coder simply encodes the possible values of the category using a dictionnary. This dictionnary is generated using the Huffman algorithm with a ternary tree. As described earlier, this will give us a dictionnary of codewords composed of digits between 0 and 2. Those codewords are then encoded into DNA using a goldman encoder with the rotating [A, T, C, G] alphabet. A rotating alphabet is an alphabet where you take out the element that you previously used, to avoid repetitions. You put the element back in the alphabet after having written a different element, meaning one step after taking it back. The order of the rotating alphabet is always the same: we don’t append the element at the end of the alphabet but at the place it was before. Having a ternary tree for the Huffman encoder was obviously a choice to be able to encode the words into a 4-element alphabet with the goldman encoder.
Value Coder¶
The value coder is the coder that is in charge of encoding the coefficient value according to the category in which it lies. It is based on the use of a codebook of codewords of fixed length. The bigger the category , the longer the codewords and the bigger the codebook. For encoding, you use the codeword that has as index the coefficient value. For decoding, you search the index of the codeword in the codebook. The codebook only changes in function of the category.
Transforms¶
Principle¶
The same kind of modularity principle has been applied when developping transforms.
Inherit from the abstract class
transforms.AbstractTransformImplement the abstract methods
mytransform.forward(inp)andmytransform.inverse(code)Both those methods only communicate with the user, the data to be encoded or decoded and the data result
Additionnal arguments can be set or accessed by implementing the
mytransform.set_state(*args)andmytransform.get_state()metodsThose arguments will be defined as attributes,
mytransform.set_state(*args)andmytransform.get_state()are a setter and a getterThe
mytransform.full_forward()ormytransform.full_inverse()methods can be overloaded to describe the full encoding or decoding process (initialisation, encoding, return of arguments)
The transforms that have already been implemented are the following:
Color Conversion Transform¶
The color conversion that we use is just a wrapper of the cv2.cvtColor function.
- class jpegdna.transforms.colortransform.RGBYCbCr[source]
RGB YCbCr color converter
- forward(inp)[source]
Wrapper method to covert RGB to YCbCr
- Parameters
inp (np.array) – Input image
- Returns
YCbCr image
- Return type
np.array
- inverse(inp)[source]
Wrapper method to covert YCbCr to RGB
- Parameters
inp (np.array) – Input YCbCr image
- Returns
RGB image
- Return type
np.array
Discrete Cosine Transform¶
The Discrete Cosine Transform that we use is just a wrapper of the scipy.fftpack.dctn and scipy.fftpack.idctn functions.
It converts from RGB with .forward() to YCbCr an back with .inverse().
- class jpegdna.transforms.dctransform.DCT[source]
For Jpeg
- forward(inp)[source]
Forward 2D DCT
- Parameters
inp (np.array) – input image block
- Returns
DCT coefficients
- Return type
np.array
- full_forward(inp, *args)[source]
Forward 2D DCT
- Parameters
inp (np.array) – input image block
norm (str) – Type of DCT (default: None)
- Returns
DCT coefficients
- Return type
np.array
- full_inverse(inp, *args)[source]
Inverse 2D DCT
- Parameters
inp (np.array) – input image block
norm (str) – Type of DCT (default: None)
- Returns
DCT coefficients
- Return type
np.array
- inverse(inp)[source]
Inverse 2D DCT
- Parameters
inp (np.array) – DCT coefficients
- Returns
image block
- Return type
np.array
Channel Sampler Transform¶
This transform contains different samplers for color channels. You sample using .forward(), it returns a tuple containgin the 3 channels with different sizes. Desampling is done using .inverse(), it gives back a 3 channel image.
To get the appropriate sampler, you instantiate the class with the name of the sample, for example ChannelSampler(sampler=”4:2:2”).
Available samplers are: "4:2:2", "4:2:0", "4:1:1", "4:4:4", "4:4:0".
- class jpegdna.transforms.sampler.ChannelSampler(sampler='4:2:2')[source]
Channel Sampler
- Variables
sampler – Sampler name (default: “4:2:2”)
- Parameters
sampler – str
- forward(inp)[source]
Sampling method
- Parameters
inp (np.array) – 3-channel image
- Returns
Tuple of the 3 sampled channels (Y not subsampled usually)
- Return type
tuple
- get_sampler()[source]
Get sampler name
- inverse(inp)[source]
Desampling method
- Parameters
inp (np.array) – Tuple of the 3 sampled channels (Y not subsampled usually)
- Returns
3-channel image
- Return type
np.array
- set_sampler(sampler)[source]
Set sampler from its name
Zig-Zag Transform¶
This transform is used to organize the DCT quantized values that are in a block in a 1-dimentional sequence of values and backwards. The forward takes a 2D array as input and outputs a 1D array and the inverse has inverse inputs and outputs.
For the inverse transform, it is required to set the expected block dimensions for the output using zigzag.ZigZag.set_state(width, height).
- class jpegdna.transforms.zigzag.ZigZag(verbose=False)[source]
Zig-zag transform
- Variables
verbose – Verbosity enabler
vmax (int) – width of the matrix
hmax (int) – height of the matrix
- Parameters
verbose – bool
- forward(inp)[source]
Transform the inp matrix into a sequence element following the zig-zag reading order
- Parameters
inp (np.array) – Input to be transformed
- Returns
Zig-zag read sequence
- Return type
np.array
- full_inverse(inp, *args)[source]
Inverse transform wrapper method
- Parameters
inp – Input to apply inverse transform
args (any) – Supplementary arguments for the inverse transforms
- Type
any
- Returns
Inverse transformed message
- Return type
any
- inverse(inp)[source]
Reconstruct matrix from a sequence of zig-zag read values
- Parameters
inp (np.array) – Sequence of values
- Returns
reconstructed matrix
- Return type
np.array
- set_state(*args, case=None)[source]
Sets the state of the transform
- Parameters
hmax –
vmax (int) –
Formatting¶
Principle¶
The formatting into oligos (small strands of size ~200 to reduce noise) of the compression result and the deformatting of those oligos to decompress an image is a totaly different layer in the general compression workflow. What this formatting needs to do is to gather all the data necessary to decode the image, meaning: the strand representing the content data, information on the frequencies that are used(from the image or from precalculated frequencies), the frequencies that should be transmitted if frequencies were computed from the image, the dimensions of the image, in the case of RGB, the sampler that was used.
Three formats of oligos will be used to format the data:
The first format is the one that will be used for the strand coming out of the compression, meaning the imag data in itself: These oligos will contain: an identifier making it possible during formatting to recognise that these are oligos related to the image data, and offset index, and a small slice of the image data strand. The offset indices indicate the order of appearance of the slices in the full data strand: during deformatting, the general data strand is reconstructing by reordering the slices according to this index.
This format is defined in the class named GeneralInfoFormatter in the format module.
- class jpegdna.format.generalinfoformatter.GeneralInfoFormatter(alpha, freq_origin, m, n, blockdims, max_cat, max_runcat, dc_freq_len, ac_freq_len, image_type, sampler, header, oligo_length=200, debug=False)[source]
Formatter for the general information related to the compression
- Variables
alpha – alpha value of the compression, if it is known
freq_origin – origin of the frequencies (“default” or “from_img”)
m – first dimension of the image
n – second dimension of the image
blockdims – dimensions of the block used for DCT
max_cat – max value for the categories
max_runcat – max value for the run/categories
dc_freq_len – codeword length for encoding the dc frequencies
ac_freq_len – codeword length for encoding the ac frequencies
oligo_length – Size of the oligos used for formatting
- colored_image()[source]
Check if image is colored or gray level
- Return type
bool
- deformat(oligos)[source]
Decoding method
- Parameters
code (list) – Formated oligos to deformat
- Returns
Deformated strand
- Return type
str
- format(inp)[source]
Encoding method
- Parameters
inp (str) – Strand to format
- Returns
Formatted oligos
- Return type
list
The second format available will concern the frequencies. Since those frequencies are number tables, they are first encoded into a two DNA strands using a codebook of fixed-length DNA codewords. One strand will concern DC categories, and one will concern AC run/categories. The values of the DC and AC categories or run/categories frequencies will be decoded from the two strands using the same codebook. The oligos for those frequencies will finally be similar to the data ones. It will contain an identifier specifying that it is a frequency oligo, an identifier stating if it is a DC or AC frequency oligo, an offset index and a samll slice of the AC or DC frequency strand.
This format is defined in the classes named GrayFrequenciesFormatter and RGBFrequenciesFormatter in the format module.
- class jpegdna.format.frequenciesformatter.GrayFrequenciesFormatter(max_cat, max_runcat, dc_freq_len, ac_freq_len, header, dc_header, ac_header, oligo_length=200, debug=False)[source]
Formatter for the gray DC and AC frequencies
- Variables
max_cat – max value for the categories
max_runcat – max value for the run/categories
dc_freq_len – codeword length for encoding the dc frequencies
ac_freq_len – codeword length for encoding the ac frequencies
oligo_length – Size of the oligos used for formatting
- decode_frequencies(strand, freq_type)[source]
Decode the frequencies using fixed-length codebooks
- Parameters
strand (str) – encoded frequency values
freq_type (str) – DC/AC frequency indicator
- Returns
list of frequency values
- Return type
list(int)
- deformat(oligos)[source]
Deformats and decodes both DC and AC frequency oligos
- Parameters
oligos (list(str)) – formatted frequency oligos for AC and DC
- Returns
decoded frequency tables
- Return type
tuple(list(int))
- deformat_frequencies(oligos)[source]
Deformat the oligos describing either the DC or the AC frequencies
- Parameters
oligos – oligos to deformat
freq_type (str) – DC/AC frequency indicator
- Returns
strand encoding the frequencies
- Return type
str
- encode_frequencies(freqs, freq_type)[source]
Encode the frequencies using fixed-length codebooks
- Parameters
freqs (list(int)) – frequency values to encode
freq_type (str) – DC/AC frequency indicator
- Returns
strand encoding the frequencies
- Return type
str
- format(inp)[source]
Encodes and formats both DC and AC frequencies
- Parameters
inp (tuple(list(int))) – frequencies for DC categories and AC run/categories
- Returns
formatted frequency oligos
- Return type
list(str)
- format_frequencies(inp, freq_type)[source]
Format the strand encoding either the DC or the AC frequencies
- Parameters
inp (str) – strand to format
freq_type (str) – DC/AC frequency indicator
- Returns
formatted AC or DC frequency oligos
- Return type
list(str)
Finally, a third type of oligo will concern one single oligo that will encode all the necessary data for deformatting and for decoding: It contains: the size of the image, the size of the blocks for the DCT, the value of the dynamic used, the length of the codewords used to encoded the frequencies in the frequency oligos. Since these informations are crucial to ensure deformattability/decodability of the data, they are encoded using barcodes, meaning that the codebooks used to encode the alpha values, … contain codewords that have an bigger edit distance. That way, if because of the noise modifies the codeword, we can try to get it back by finding the closest codeword present in the codebook.
This format is defined in the class named DataFormatter in the format module.
- class jpegdna.format.dataformatter.DataFormatter(header, oligo_length=200, debug=False)[source]
Formatter for the strand coming out of the compression algorithm
- Variables
oligo_length – Size of the oligos used for formatting
- deformat(oligos)[source]
Methods deformatting the oligos and rebuilding the data strand
- Parameters
oligos (list(str)) – data oligos
- Returns
data strand resulting from compression
- Return type
str
- format(inp)[source]
Method formatting the data strand into oligos
- Parameters
inp (str) – strand resulting from the compression algorithm
- Returns
list of formatted oligos of size self.oligo_length
- Return type
list(str)
In addition to the formatting that was described earlier, all the oligos will contain a sense nucleotide at each end indicating in which sense it should be read, an parity check that will act like a checksum, containing four oligos, each of them checking the parity of one base (A, T, C, G). They will also contain an identifier that is unique for every image in the oligo pool. Finnaly for real usecase applications, one can need to add primers for sequencing machines. We already included in the code the case where people want to use illumina primers.
All this formattingalgorithm has been integrated in the codecs. A boolean parameter called formatting is used at initialization of the codec to specify if one wants to use formatting or not.
